Ultimo Aggiornamento 11 Febbraio 2016
La fenilchetonuria è una malformazione congenita causata da una mutazione in un gene che aiuta a creare l’enzima necessario per abbattere fenilalanina.
Gli aminoacidi sono i mattoni delle proteine, ma lafenilalanina in eccesso può causare una varietà di problemi di salute. Le persone affette da fenilchetonuria (PKU) – neonati, bambini e adulti – devono seguire una dieta che limiti la fenilalanina, che si trova principalmente negli alimenti che contengono proteine.
Sintomi
I neonati affetti da fenilchetonuria inizialmente non hanno alcun sintomo. Senza trattamento, però, i bambini di solito si sviluppano segni di PKU in pochi mesi. I sintomi possono essere lievi o gravi e possono comprendere:
- Ritardo mentale
- Problemi comportamentali o sociali
- Convulsioni, tremori alle braccia e alle gambe
- Iperattività
- Crescita stentata
- Eruzioni cutanee (eczema)
- Testa piccola (microcefalia)
- Problemi nella pigmentazione ( pelle molto chiara occhi chiari)
- Un odore di muffa nella pelle o nelle urine, causata dalla fenilalanina in eccesso
Diversi stadi di gravità
La forma più grave della malattia è conosciuta come classica PKU. I bambini affetti da PKU non trattata sviluppano solitamente un ritardo mentale permanente.
Forme meno gravi di PKU hanno un minor rischio di danni cerebrali significativi, ma la maggior parte dei bambini con queste forme della malattia necessitano ancora di una dieta speciale per prevenire il ritardo mentale e altre complicazioni.
Gravidanza e PKU
Una donna che ha la PKU e rimane incinta è a rischio di un’altra forma della malattia denominata fenilchetonuria materna. I medici raccomandano che chiunque abbia la PKU debba seguire la dieta a basso contenuto di fenilalanina per tutta la vita.
Sebbene i bambini nati da madri con alti livelli di fenilalanina possano avere complicazioni alla nascita, la maggior parte in realtà non ereditano la PKU. Tuttavia, questi bambini hanno un alto rischio di nascere con:
- Ritardo mentale
- Microcefalia
- Difetti cardiaci
- Basso peso alla nascita
- Problemi comportamentali
Modalità di trasmissione autosomica recessiva
Il gene difettoso contiene le istruzioni per creare un enzima necessario per elaborare l’aminoacido chiamato fenilalanina. Gli aminoacidi sono i mattoni per le proteine. In una persona con PKU, questo gene è difettoso, e causa una carenza completa o quasi completa dell’enzima. Senza l’enzima necessario per elaborare la fenilalanina, un accumulo pericoloso di questo aminoacido è in grado di svilupparsi quando una persona mangia alimenti che sono ricchi di proteine, come latte, formaggio, noci o carne. Questo può portare a gravi problemi di salute.
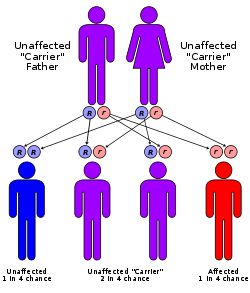
Per ereditare la PKU, sia la madre che il padre devono avere e trasmettere il gene difettoso. Questo modello di ereditarietà è chiamata autosomica recessiva. E’ possibile che un genitore abbia il gene difettoso, ma non abbia la malattia. Questo si chiama vettore. Il più delle volte, la PKU è passata ai bambini dai genitori che sono portatori della malattia, ma non lo sanno.
Fattori di rischio
Entrambi i genitori devono passare il gene mutato della PKU per sviluppare la malattia. Se solo un genitore ha il gene della PKU, non c’è rischio di trasmissione.
Il difetto genetico si verifica soprattutto nelle persone provenienti da Europa e Stati Uniti La malattia è molto meno comune negli asiatici e latini.
Figli di madri che hanno la PKU, ma che non hanno seguito la dieta corretta durante la gravidanza possono essere colpiti da gravi conseguenze date dell’alto livello di fenilalanina nel sangue della madre.
Complicazioni
La PKU non trattata può portare a:
- Danni cerebrali irreversibili e marcato ritardo mentale entro i primi mesi di vita
- Problemi comportamentali e convulsioni nei bambini più grandi
Diagnosi
Semplici analisi del sangue nel neonato identificano quasi tutti i casi di PKU.
Se si dispone di PKU o una storia familiare di PKU, il medico può raccomandare test di screening prima della gravidanza. E’possibile identificare i portatori sani di PKU attraverso un esame del sangue.
Trattamenti e cure
Il trattamento principale per la fenilchetonuria è una rigorosa dieta a basso contenuto di fenilalanina, che si trova soprattutto nei cibi contenenti proteine. I medici raccomandano di attenersi alla dieta per tutta la vita.
La quantità sicura di fenilalanina è diversa per ogni persona. Il medico stabilirà un ammontare sicuro attraverso una revisione periodica della dieta, tabelle di crescita e livelli ematici di fenilalanina. Esami del sangue frequenti monitoreranno i livelli di PKU che variano nel tempo, specialmente durante la crescita e la gravidanza. In generale, l’idea è quella di consumare solo la quantità di fenilalanina che è necessaria per la normale crescita e per i processi del corpo, ma non di più.
Quali alimenti evitare
E’ fondamentale evitare che tutti i cibi ad alto contenuto proteico, tra cui:
- Latte
- Uova
- Formaggio
- Noci
- Germogli di soia
- Fagioli
- Pollo
- Bistecca di manzo e prodotti simili
- Carne di maiale
- Pesce
- Cioccolato, caramelle
- Piselli
- Birra
I bambini e gli adulti dovrebbero evitare anche molte bevande dietetiche, e farmaci a base di aspartame (NutraSweet, Equal).
Adulti e bambini affetti da PKU dovrebbero anche limitare porzioni di:
- Frutta
- Verdure
- Dessert
E’ fondamentale ricordare che il troppo di ogni cosa a volte può essere dannoso. Anche se si sta mangiando un cibo consentito, mangiarne troppo può essere pericoloso. Considerare la quantità totale di fenilalanina in tutti gli alimenti al momento di pianificare la dieta.
Farmaci
La Food and Drug Administration (FDA) ha approvato il farmaco dicloridrato (Kuvan) per il trattamento della PKU. Agisce aumentando la tolleranza alla fenilalanina. Il farmaco deve essere utilizzato in combinazione con una dieta. ma non funziona per tutti. Nell’approvare il farmaco, la FDA ha disposto che gli studi continuino perché non ci sono studi a lungo termine sull’efficacia del farmaco e la sicurezza del suo impiego.
Consigli
Tenere un diario alimentare
Se voi o il vostro bambino state seguendo una dieta a basso contenuto di fenilalanina, avrete bisogno di registrare le quantità di fenilalanina mangiata ogni giorno per essere sicuri di attenersi alle linee guida dietetiche personalizzate consigliate dal dietista.
Per fare questo, utilizzare un diario alimentare o un programma per computer che elenca la quantità di fenilalanina negli alimenti.
Basso contenuto di proteine
Comprare alcuni dei molti prodotti creati a basso contenuto proteico, come riso, farina e pane, che sono disponibili presso i rivenditori di specialità alimentari.
Questi prodotti offrono una certa varietà, e consentono alle persone con PKU di mangiare pranzi e cene che si avvicinano maggiormente a quelli normali.
Fenilchetonuria. Genetica e riferimento. http://ghr.nlm.nih.gov/condition=phenylketonuria. Letta 6 ottobre 2011.
Blau N, et al. Fenilchetonuria. The Lancet. 2010; 376:1417.
Bodamer OA. Panoramica di fenilchetonuria. http://www.uptodate.com/home/index.html. Letta 6 ottobre 2011.
PKU (fenilchetonuria). March of Dimes. http://www.marchofdimes.com/baby/birthdefects_pku.html. Letta 6 ottobre 2011.
Dieci Hoedt AE, et al. Crescere un bambino con galattosemia o fenilchetonuria: Implicazioni per la salute la qualità della vita. Giornale di malattie ereditarie metaboliche. 2011; 34:391.
Van Spronsen FJ et al. Grandi aminoacidi neutri nel trattamento della PKU: Dalla teoria alla pratica. Giornale di malattie ereditarie metaboliche. 2010; 33:671.



